Current state of cryo-EM maps and models
Cryo electron-microscopy is a method to determine structures of macromolecules, such as proteins, enzymes and viruses. The repositories for the models and maps are the EMDB and the PDB, respectively. Recent technological and methodological advances have led to more and more successful cryo-EM structure determination experiments. The figures below show the evolution of some properties of deposited models and maps over time (last update: 04/2024).
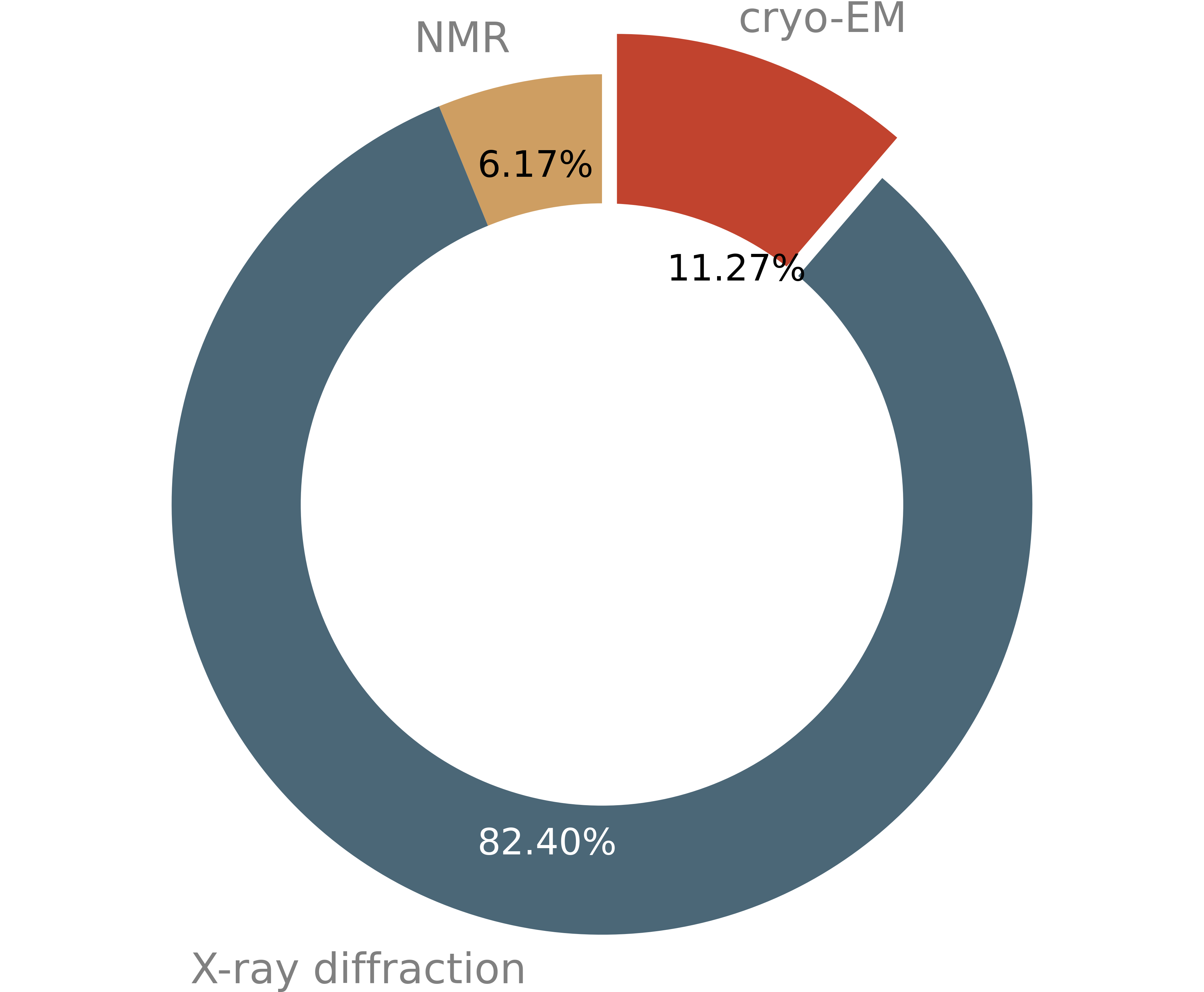
After X-ray crystallography and nuclear magnetic resonance (NMR), cryo-EM is now the third most used method for macromolecular structure determination.
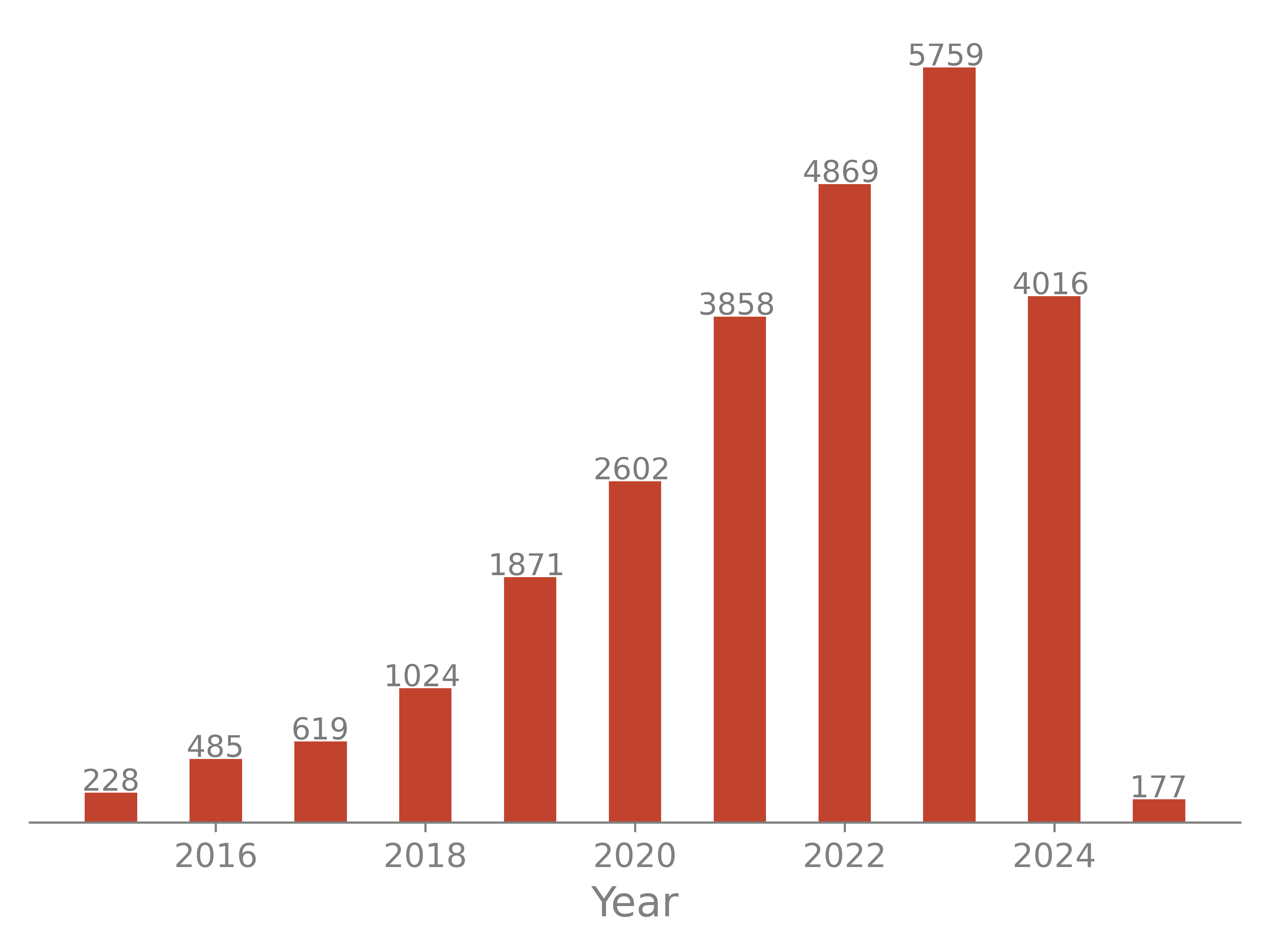
The number of models determined with the cryo-EM method increases steadily in the past years.
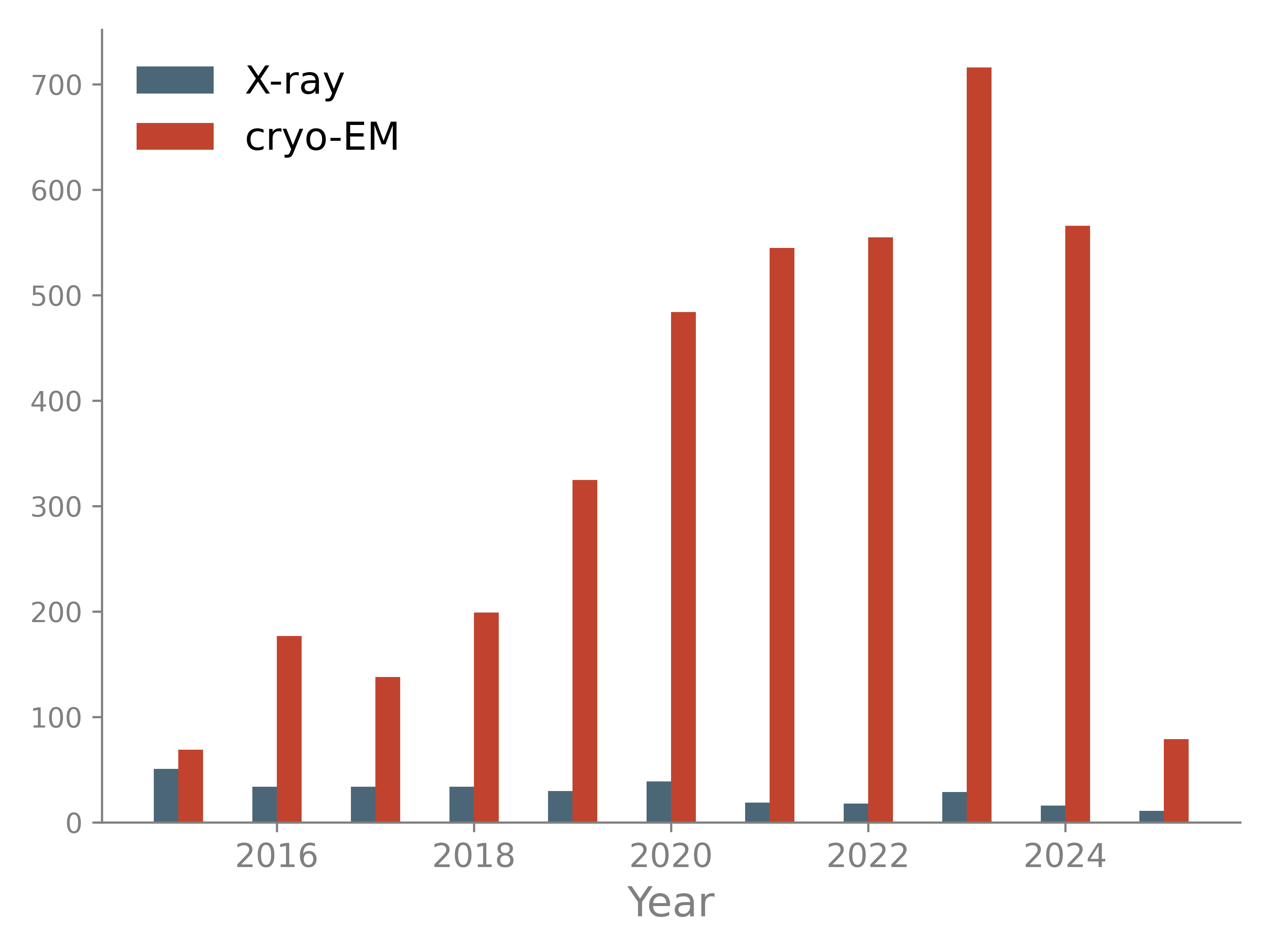
Most large structures are now determined with cryo-EM while the number of large X-ray models decreased in the past years. Large models pose several challenges: they require robust automation procedures to avoid manual analysis of thousands of residues; associated maps may have low resolution and are therefore difficult to interpret; time for computations may increase significantly.
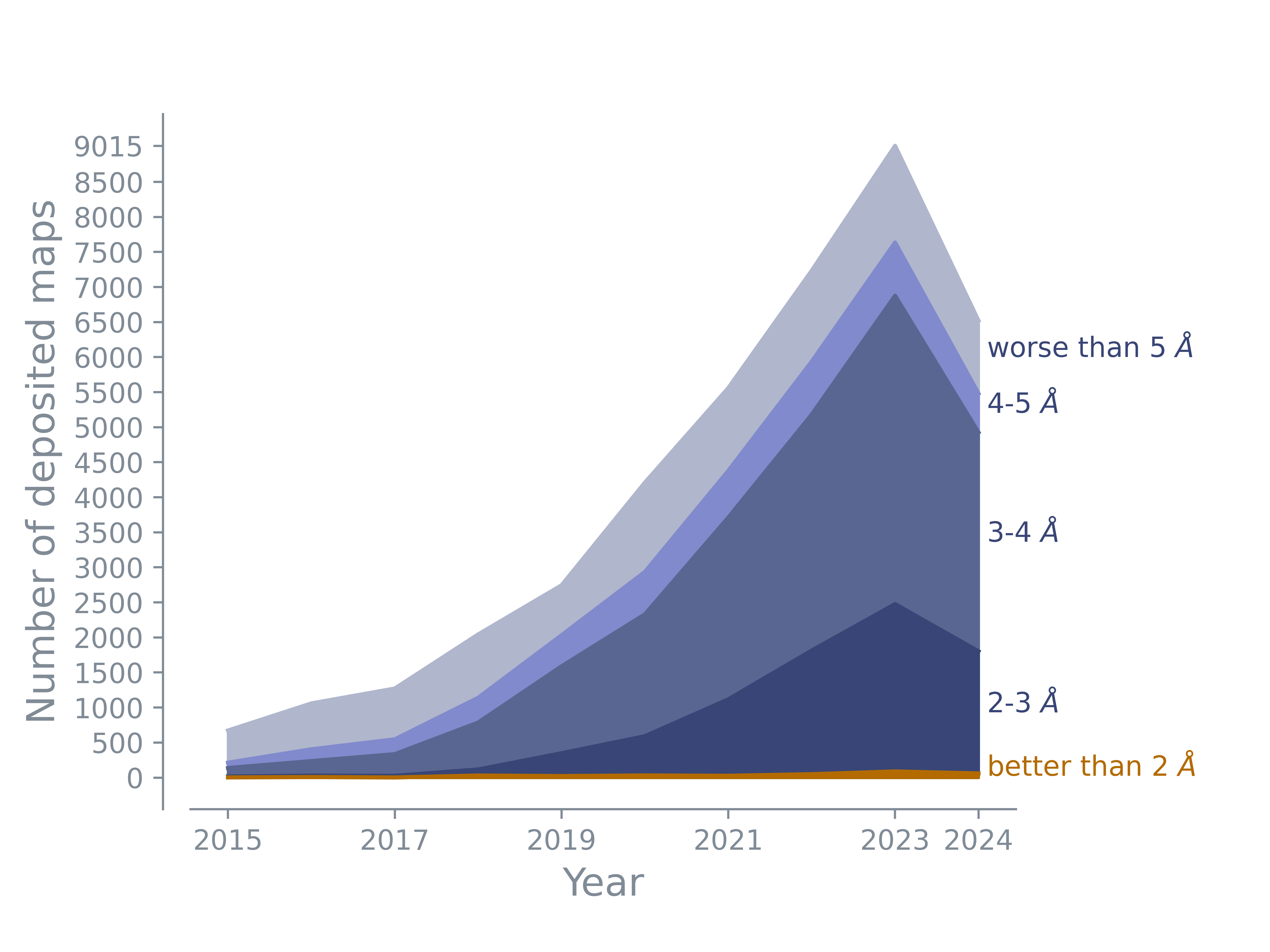
The majority of deposited maps has a resolution between 3 and 5 Å. The number of maps with resolution worse than 5 Å maps remains constant. There are more and more maps with resolutions better than 3 Å or even 2 Å. The number in this plot shows the number of maps; the map may or may not have an associated model.